时间:2024-05-16 10:36来源:上海交通大学医学院附属作者:王依柔 等
患儿,男,汉族,3 岁 3 个月,因身材矮小伴发育落后就诊。
患儿体质量 9 kg,身高 74 cm,均位于同年龄、同性别儿童 P3 以下;现刚会独立行走,有时仍需搀扶,尚不会说话(只会发无意义的单字)。
患儿系 G2P2,孕 40 周出生,出生身长41 cm(男童出生身长-3SD为 45.2 cm),出生体质量 1760 g(男童出生体质量-3SD 为 2.26 kg),为足月小样儿。
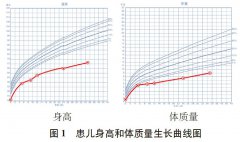
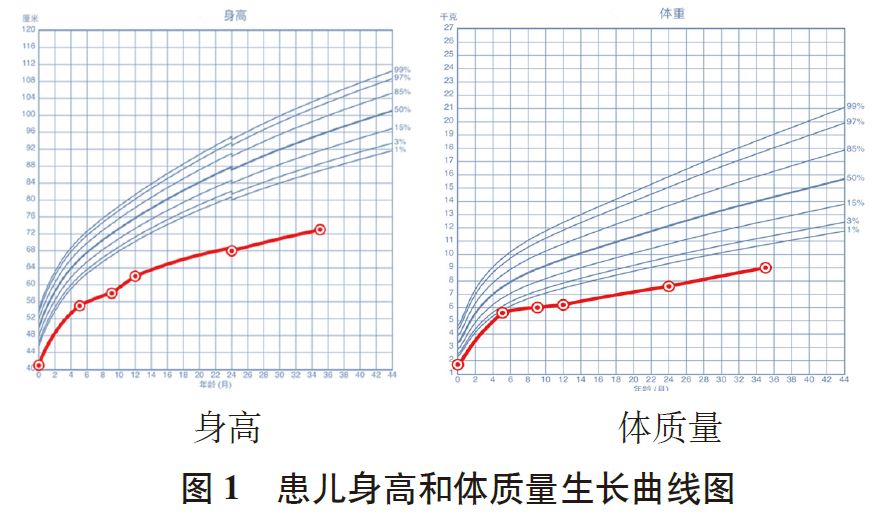
出生后母乳喂养,婴幼儿期生长发育落后,根据随访记录,将患儿生后 5 个月、9 个月、1 岁、2 岁的身高、体质量,绘制生长曲线图显示,身高、体质量始终位于同年龄、同性别儿童生曲线图的P3 线以下(图1)。
于 2015 年 11 月曾行左侧隐睾手术。
父母非近亲婚配,家族中无遗传代谢类疾病史;有一姐姐,4 岁 7 个月,生长发育正常。
神清,精神反应可,浅表淋巴结无肿大;前发际偏高,眼距稍宽,鼻梁塌陷、宽鼻尖,下唇薄,下颌小,耳位低,短颈;双手手指粗短,皮肤稍黑,双侧掌纹呈“通贯手”;心肺腹无异常;腹股沟见隐睾术后疤痕,小阴茎;神经系统检查未见异常。
肝肾功能、甲状腺功能、性激素等均无异常。
两次查类胰岛素样生长因子(IGF- 1)分别为 49 、54.1 ng/mL。
皮质醇 7.5 ng/mL。
促肾上腺皮质激素 21.10 pg/mL。
串联质谱检查所有氨基酸和酰基肉碱谱均无显著异常。
外周血染色体核型分析(550带,G带)见46 条染色体,数目正常,性染色体为XY,染色体结构未见异常(图3)。

7 月龄时 GESELL 评分:运动能 DQ 粗运动 82 (相当于 4 个月)、细运动 56(相当于 3 个月),应物能 DQ 43(相当于 2.3 个月),言语能 DQ 69(相当于 3.7 个月),应人能DQ56(相当于 3个月),均落后于正常。
头颅 MRI 示轻度脑室周围白质软化,胼胝体偏薄,双侧额部脑沟增宽,双侧乳突积液。
阴囊 B 超示双侧睾丸在位(术后)。
心脏彩超示卵圆孔未闭。
腹部 B 超示肝脾、双肾、均偏小。
1 岁 11 月龄骨龄平片示骨龄落后 5~8 个月。
经父母知情同意,行细胞遗传学检查并完善全基因芯片扫描。使用 SurePrint G3 Custom CGH+SNP(4*180 K) 芯片进行全基因组扫描,发现患儿 1 号染色体 q24.3~q25.3 区域存在一段大小为 14615 kb 的杂合缺失(图4)。

矮小症是儿科常见疾病,是指与同地区、同年龄、同性别正常儿童比较,身高低于正常儿童生长曲线的 P3。引起儿童矮小的病因繁多,与遗传、营养、环境、精神心理因素、宫内发育迟缓、下丘脑-垂体-胰岛素样生长因子 1 轴异常、染色体畸变、全身性慢性疾病、遗传代谢病以及内分泌激素等关系密切。在患儿出生早期寻找致生长迟缓病因是诊疗的重点。
目前为止,对于1 号染色体长臂缺失的报道仍极为罕见。1980 年 Schinzel 等根据缺失部位将 1 号染色体缺失分为 3 种类型:1 q 近端缺失(1q21~22 → q25)、1q 中间缺失(1q24~25 → q32)以及 1q 远端缺失(1q42 → 末端)。其中 1q 中间缺失型已报道 40 余例,但缺乏共同的特征性表型。近年来,Burkardt 等提出 1q24 q25 微缺失综合征的概念,即伴有 1q24 q25 缺失的患者,具有共同的特征性临床表现,包括出生前及出生后的生长发育迟缓、短指畸形以及特殊面容(上眼睑肥厚、异常小耳朵、短鼻伴球状鼻尖、帐篷样上唇以及小颌畸形等)。随着基因诊断技术的发展,对染色体畸变的诊断更为准确,这类患者的报道也逐渐增多。
本例患儿出生时及出生后身高、体质量均低于同年龄、同性别儿童-3SD,语言、行为以及智力发育均明显落后,还伴有隐睾、心血管系统畸形、通贯手、小颌畸形等。考虑到染色体异常是引发先天畸形、精神疾患以及智力缺陷的常见原因,故对患儿外周血染色体核型进行分析,但结果为阴性。再行全基因芯片检测,发现 1 号染色体 q24.3 ~ q25.3 区域存在一段大小为14 615 kb(约14.3 Mb)的杂合缺失,该区域包含多个致病基因如ABL2、ACBD6、DNM3、CENPL、DARS2、LHX4、TNFSF4、PRDX6、XPR1等,其中LHX4基因位于 1q25,在调控运动神经元的支配以及促进垂体发育过程中起作用。曾有报道 1 例家族性 LHX4杂合突变,其表型特征为身材矮小,垂体前叶激素严重缺乏,垂体和脑后部发育缺陷以及蝶鞍畸形,缺失这一区域所导致LHX4单一不足可造成儿童生长激素的缺乏。IGF- 1 的生理作用是促进骨骼细胞的分化和增殖,介导生长激素对生长的促进作用。本例患儿IGF- 1 始终处于同年龄层的低水平,提示可能存在生长激素缺乏,但其生长激素水平还需进一步的生长激素激发试验明确。另一个可能导致该综合征的基因为DNM3,其是导致身材矮小、小头畸形以及神经发育障碍的主要基因之一。DNM3 由 18 个外显子组成,编码含 702 个氨基酸的蛋白 dynain-3。该蛋白在成人的大脑和脊髓中高表达,可与突触后 Homer 蛋白结合形成 Shank-Homer 复合物,起到促进谷氨酰胺能神经元成熟以及增加突触强度的作用。最新报道显示,在 DNM3 的内含子中有 7 kb 的反义转录本编码 2种小分子RNA(miR199、miR214),在与面部颅骨发育相关的间质和软骨膜中高表达。研究发现,敲除miR199+214 的小鼠,虽然 dynain-3 蛋白的功能可以正常表达,小鼠仍表现为喂养困难、生长停滞、颅面发育不全以及骨质疏松等。本例患儿手掌X片示手小、短指和宽指,临床可见身材极度矮小、下颌小、眼距宽,在一定程度上反映了骨骼的改变。此外,CENPL基因位于 1q25.1,编码着丝粒蛋白L,在着丝粒发挥正常功能和有丝分裂过程中起重要作用。CENPL基因缺失可能是导致伴有 1q24q25 缺失患者生长发育迟缓和小头畸形发生的原因之一。
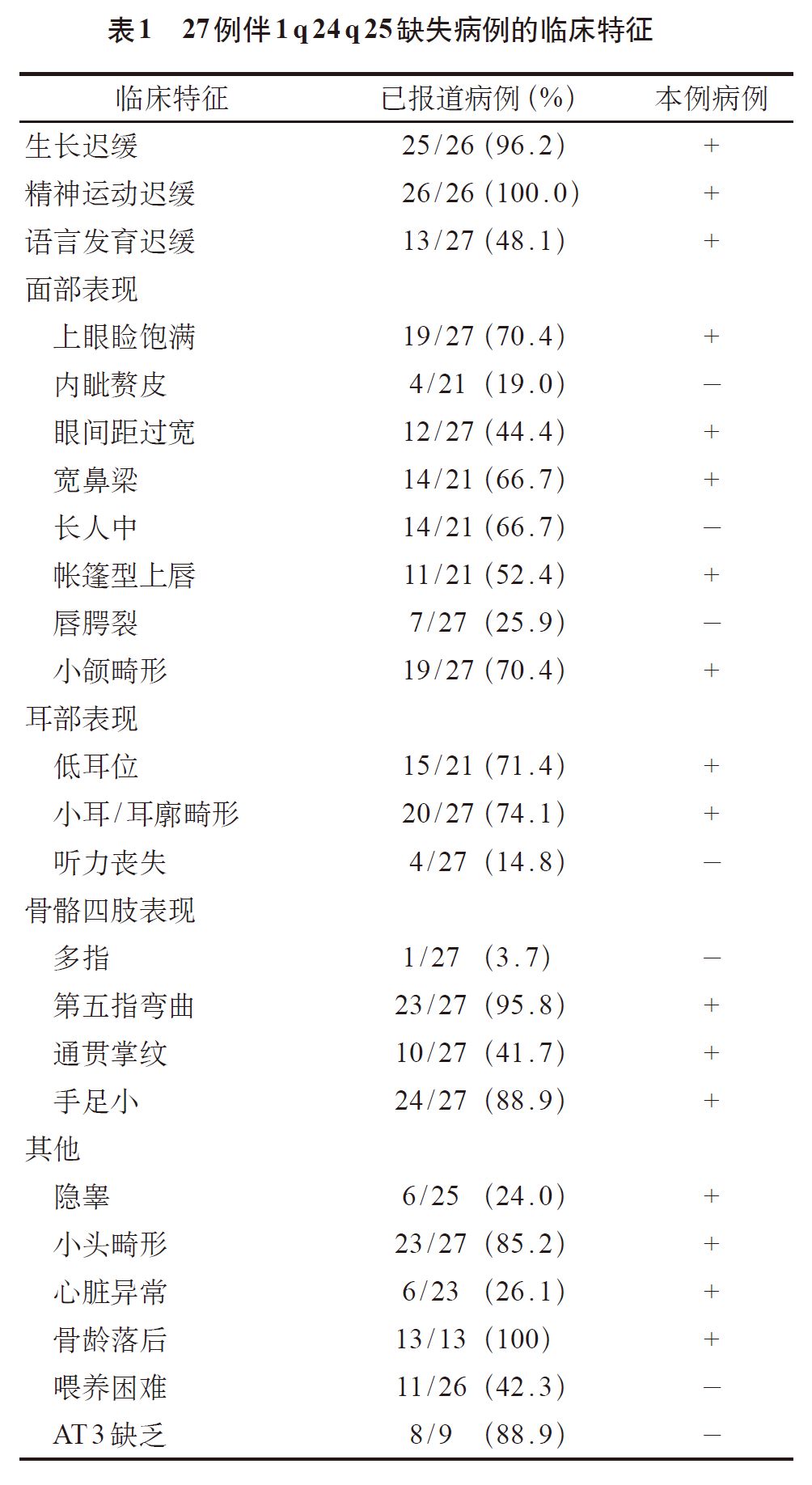
通过回顾以往病例并进行归纳分析发现,在 1 号染色体长臂缺失中,伴 1q24 ~ q25 缺失的患者拥有类似的表型。本例患儿的临床特征与已报道过的病例基本相符(表1),提示 1 号染色体q24.3 ~ 25.3 区域这段 14 615 kb 的杂合缺失可能为该患儿严重矮小、特殊面容、发育迟缓以及多发畸形的重要致病因素。
在本例患儿的诊治过程中,首先选择染色体核型分析,因其费用较低,可通过分析细胞内染色体的长度、着丝点位置、臂比等特征,并根据染色体的结构和数目的变异来判断疾病病因。但染色体核型分析存在局限性,无法检测出微结构异常(分辨率> 5 Mb),对于大片段结构异常的检出也受主观判断和个人经验的影响。本例患儿片段缺失的染色体为 1 号染色体,1 号染色体为人类染色体中最大的一条,包含的基因数目最多,在核型分析中,染色体发生缠绕等客观因素也可造成检查结果的误差。全基因芯片分析与染色体核型分析相比,不需要进行细胞培养,分辨率高出近千倍,几乎可用于任何组织的 DNA 分析。全基因芯片分析可在全基因组范围内同时检测到多种染色体不平衡导致的遗传性疾病,可检测到染色体的微重复和微缺失,并能客观准确地界定拷贝数变异的区间及大小。
临床工作中以矮小伴多发畸形就诊的患者需考虑到染色体畸变的情况,应通过积累病史资料、临床表型等,进行系统详细的临床分析,同时选择合适的遗传学检测方法,才能得到更为全面和准确的诊断。